INBORN ERRORS OF METABOLISM (What they are and how they affect unborn children)
Introduction and General Overview
It takes a whole lot of metabolic activities for the human body to survive. From breathing to eating, walking and every other human activity to happen, the body must metabolize the macromolecules which include carbohydrates, proteins, lips, vitamins and nucleic acids. Almost every step in a metabolic pathway is catalyzed by one or more enzyme. These enzymes act as the driving force which facilitates the smooth running of the biochemical pathways. If there is a defect in any of the enzymes of a metabolic pathway, it may result in an inborn error of such metabolic pathway.

Therefore, inborn errors of metabolisms are defined as rare genetic disorders caused by a defect in the activity or quantity, or total absence of one or more enzymes of a particular metabolic pathway. Inborn errors of metabolism affect the body at genetic levels; hence they often result in congenital disorders (inherited disorders). Majority of inborn errors of metabolism are caused by a defect in a single gene which codes for the production of a particular enzyme which converts various substrates into products.
In simpler terms, the defect or absence of an enzyme which is required to convert a substrate into a product, leads to the accumulation of toxic substances which can interfere with normal bodily functions, or hinder the synthesis of important compounds.
In this series, we will talk about different types of inborn errors of biochemical metabolism.
Classification of Inborn Errors of Metabolism
Every metabolic pathway has its own enzymes; this makes it susceptible to disorders. From the glycolytic pathway to the urea cycle, every pathway can suffer loss of disorder of one or more enzymes, and end up producing toxic metabolite or deficient amount of essential products. Congenital metabolic disorders are therefore classified into the following:
- Disorders of carbohydrate metabolism
- Disorders of amino acid metabolism
- Disorders of fatty acid metabolism and mitochondrial oxidation
- Disorders of purine and pyrimidine metabolism
- Disorders of organic acid metabolism
- Disorders of pophyrin metabolism
- Disorders of steroid metabolism, etc.

I stated earlier, in this series, we would be talking about these disorders one after the other. For the sake of this article, let’s focus on inborn errors of carbohydrate metabolism.
INBORN ERRORS OF CARBOHYDRATE METABOLISM
The body metabolizes carbohydrates through a series of anabolic and catabolic pathways. Carbohydrate metabolism commences in the small intestine, where simple sugars (monosaccharides and disaccharides) are absorbed into the intestinal walls.
Inborn errors of carbohydrate metabolism are genetic defects in any enzyme involved in any of the pathways of carbohydrate metabolism.
Inborn errors of carbohydrate metabolism are thus classified into
- Defects of galactose metabolism
- Defects of glycogen degradation
- Defects of pyruvate metabolism
- Defects of fructose metabolism
- Defects of gluconeogensis
GALACTOSE METABOLISM AND GALACTOSEMIAS
Galactose is mainly obtained from lactose through the action of ß-galactosidase (lactase). A little amount also comes from the degradation of glyco proteins, Glycolipids and other complex carbohydrates in the intestine. Galactose must be metabolized into Glucose-6-phosphate before it can be used for energy production in the gycolytic pathway. This conversion is done through a series of reactions called the Leloir pathway. The pathway is named after Luis Federico Leloir, the first scientist who described the overall process of galactose metabolism.
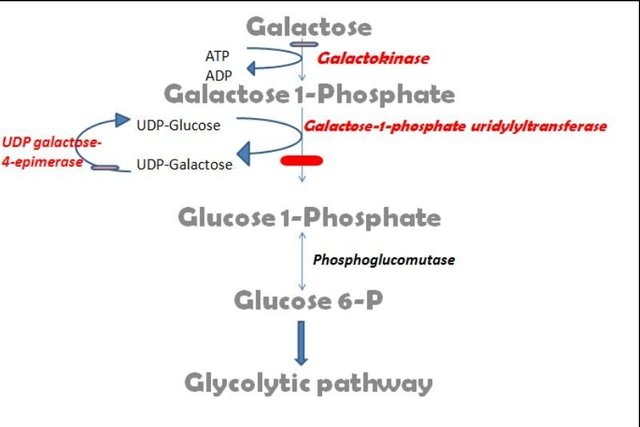
(Image designed by me)
Firstly, galactose is phosphorylated to Galactose-1-phosphate (G-1P) by the enzyme galactokinase. Galactokinase is encoded by the GALK1 gene, which spans 7.3 kbps. It has 8 exons which is a 392 amino acid protein, while the second isoform is a 270 amino acid protein.
Galactose-1-phosphate is further converted to UDP-galactose through a reaction with UDP-glucose to produce Glucose-1-phosphate and UDP-galactose. This reaction is catalyzed by galactose 1-phosphate uridyltransferase (GALT). The gene encoding for the GALT enzyme is located on chromosome 9p13.3; it has 11 exons which produces two alternatively spliced mRNAs, which encodes two different isoforms. The first isoform is a 379 amino acid protein.
Glucose-1-phosphate is then converted to Glucose-6-phosphate by phosphoglucomutase.
Classical galactosemia results from a mutation in the gene which codes for the synthesis of GALT. It is also called Type 1 galactosemia. This disorder is an autosomal recessive disorder which occurs once in 60,000 births.
A second type of galactosemia called Type 2 galactosemia is caused by the deficiency of galactokinase (GALK1). This defect is rarer than type 1 galactosemia, as it occurs in 1:100,000 births.
Type 3 galactosemia arises due to a defect in UDP-galactose-4-epimerase.
All three types of galactosemia causes the toxic accumulation of Galactose, galacitol and Galactose-1-phosphate in the liver, the nerves, lens and kidney tissues results in a galactosemia and galactosuria, which comes with physiological implications such as:
Jaundice
Hepatic insufficiency
Mental retardation
Poor growth
Cataract
Vomiting
Diarrhea
DIAGNOSIS AND TREATMENT OF GALACTOSEMIA
Diagnosis
Galactosemia tests are done with urine or blood sample (collected from the heels of the child). It tests for the presence of the three enzymes needed for metabolism of galactose. Galactosemia patients must be deficient in at least one of the enzymes, which is the reason for the accumulation of galactose in urine or blood.
Screening for galactosemia can be done prenatally by chorionic villus sampling. New born screening (NBS) is also very necessary as it can detect galactosemia before the child is breastfed or given any fomular that contains lactose. Two NBS screening methods are used in testing for galactosemia- the Hill test and Beutler’s test. Galactosemia positives are made to undergo another test called the Florida test
Treatment
The only available treatment option is the complete elimination of lactose containing food and any other sources of galactose from the diet. Parents must ensure that they read the label of all formulas to avoid feeding the child with any diet that will deposit galactose in the child’s body. Therefore, infants with galactosemia cannot be breastfed because breast milk contains lactose. Because of this, such infants are fed with soy milk, other milk-free formula and a protein hydrolysate formula called Nutramigen.
Long-term Complications
Despite early diagnosis and withdrawal of food sources of galactose and lactose, people with glycosemia still experience long-term health complications like
Learning disability
Speech difficulties
Tremor
Diminished bone density
Dysmetria
Premature ovarian failure
Ataxia
In the next episode of this series, we will continue our discussion by looking into other inborn errors of carbohydrate metabolism.
REFERENCES
Medical Genetics Lecture Notes
