Pin1 - Using Cystallography & NMR structures in Pymol to unveil a proteins function & structure
Pin1 - Using Crystallography & NMR structures in Pymol to unveil a proteins function & structure
Hello steemians, I got something interesting to share today. It regards Pin1, an important protein for cell division, cancer suppression and many other bio interactions and how we model the proteins. The post will focus on how I use or could use PyMol to render proteins and draw conclusions about their structure and functions in vivo.
I will also present some short commands usable in PyMol to render proteins, or general guidance rather, since much have been built in to the GUI now and show that crystallography and Nuclear magnetic resonance spectroscopy is great combo. It is really easy to use this program and already determined structures from The Protein Database PDB, can be imported and turned into great pictures or moving pictures / movies of molecules.
After that you ´ve read this post, you can try to import any protein from https://www.rcsb.org/ and just color and represent them however you like, don't miss out!
Pin1 is known to regulate mitotic checkpoints by interacting with NIMA, and bind to with high specificity to the sequence P.Thr/P.Ser-Proline. This means phosphorylated serine or threonine followed by a proline residual.
NIMA: "never in mitosis A-related kinase", is a protein that never is involved in mitosis haha, yeah the names are hilarious from time to time.
Another fun example is JAK - "just another kinase" or the more annoying LRP - "Low density lipoprotein receptor related protein", but at least it makes understanding the functions of these proteins easy, or like with NIMA where it is active. Whoever named these great work!
Pin1 also regulates p53 activity, a known tumor suppression protein, needs to be fully phosphorylated, at 3 positions to be activated by Pin1 and suppress tumor growth or proliferation rather. This study will show why the phosphate, especially at the serine position on the target peptide(part of protein, or amino acid sequence in this case) is so important for binding.
Some background.
Peptidyl-prolyl cis/trans isomerases in general catalyzes the cis/trans isomerization of proteins and thereby activating them. The WW domain recognizes, phosphorylated ser/thr-pro motifs to both via WW domain for specificity and the PPiase domain for catalytically activity. It is directly dependent on the post translational modification, phosphorylation. E.g. Tumor suppressor 53.
The conformational regulation catalyzed by this PPIase has a profound impact on key proteins involved in the regulation of cell growth, genotoxic and other stress responses, the immune response, induction and maintenance of pluripotency, germ cell development, neuronal differentiation, and survival. This enzyme also plays a key role in the pathogenesis of Alzheimer's disease and many cancers. Multiple alternatively spliced transcript variants have been found for this gene.[provided by RefSeq, Jun 2011][1]
PyMol & Pin1´s structure
PyMol is a free to use, open source program running on python code. There is a license you can buy, but you can just as well compilate the program you self to enjoy the full version. The limited version is good enough and I decided to use functions and commands that are available in this free version, should anyone want to try it out. I downloaded Pymol to my laptop and did all the following work in free mode.
Leave a comment if you want more guidance with pymol, it is a great software for chemists, biologists and off curse biochemists. It is also a great tool for creating molecule & protein representations for any blogger in the field.
https://pymol.org/2/
Just hit the download button for the appropriate op, and install it where you like.
Structure WW - specificity filter.
WW domains are found in many proteins and functions as the high affinity part, that binds to certain proteins. The PPias region will be pulled by the linker in proximity to the protein and perform its cis-trans isomerization. There are different WW groups and inside the same groups, there are little conversation of amino acids. This points to the fact that the WW domain more works like a flexible platform to create different recognition domains. The WW domains all have to thryphtopahnes (W) which has given it the name WW doimain. The only fully conserved residual is arg17 which I will prove, is very important for recognition of the phosphate modification sitting on the peptides serine(SEP). I will also show that a proline is important at the position after, using different crystal & NMR structures in PyMol. I will try to make a comprehensible guide, of how I am rendering the protein, and what conclusions can be drawn from them.
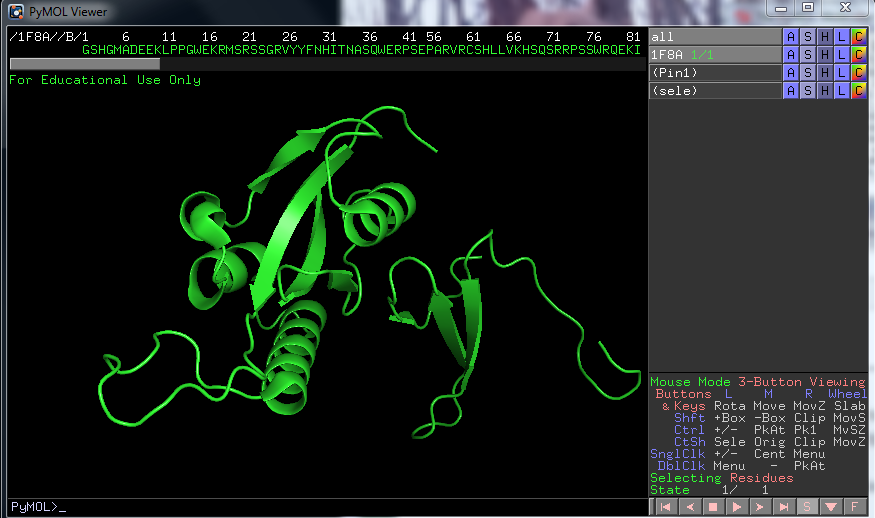
In the command line type: Fetch 1F8A[2], and a crystallography structure of Pin1 will be loaded.
To start out, in the right bottom corner, there is an button labeled [S], press it to show the full amino acid sequence.
Then I would look in the top right part, where the file name is displayed and commands buttons next to it, press "the H on the file called ALL and chose hide everything"
Now mark the entire amino acid sequence, which is now displayed at the top and chose show as cartoon, the marked part will be under the section called all in the list, click S chose cartoon.
To start naming and rendering different parts, mark the interesting part in the amino acids sequence. You just marked all amino acids, click the entry under all, named sele, and rename it to Pin1,Press the A next to the name sele to find the rename option, now we will be at this step:
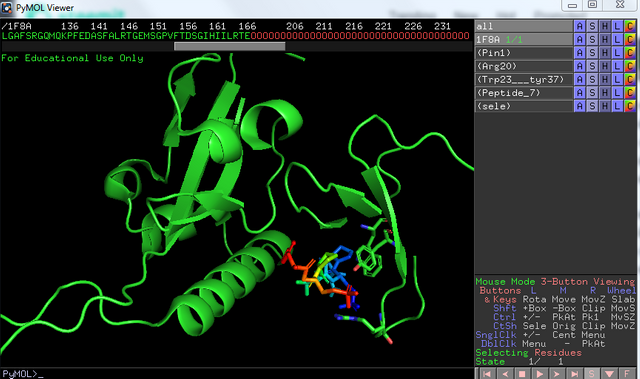
Now we can mark new amino acids in the sequence at the top, I will start with marking Tyr / Trp at positions 26 / 37 in this file. I will again look at the listed entry "sele" press the A button and choose rename - to trp / tyr 26/ 37. I will press the S on the new entry called Trp / Try 26 / 37 and chose show as sticks to get a clear view of the amino acid structure.
I will do the same with R- arginine at position 20, name it Arg20 and show it as sticks.
This is a crystal structure, crystallized with a peptide, which is shown after all the 00000 in the sequence, the 0´s are crystal waters for that matter, and can also be shown next to the protein should we want to. Mark the Y SEP PT SEP PS sequence and name it peptide or what you like to call it, show as sticks, color by b-factors and show valence. Now we are here:
The B-factor colors are more than just a color system. We have a crystal structure, which prevents the protein from moving. We also freeze the sample with liquid nitrogen, probably under 200K which will further limit movement.
The colors here range from red = high thermodynamic disorder toward blue = low values. It does not correspond to position errors directly, which some might think. It means that that part in red is capable to move, and/or are bound less rigidly to the protein.
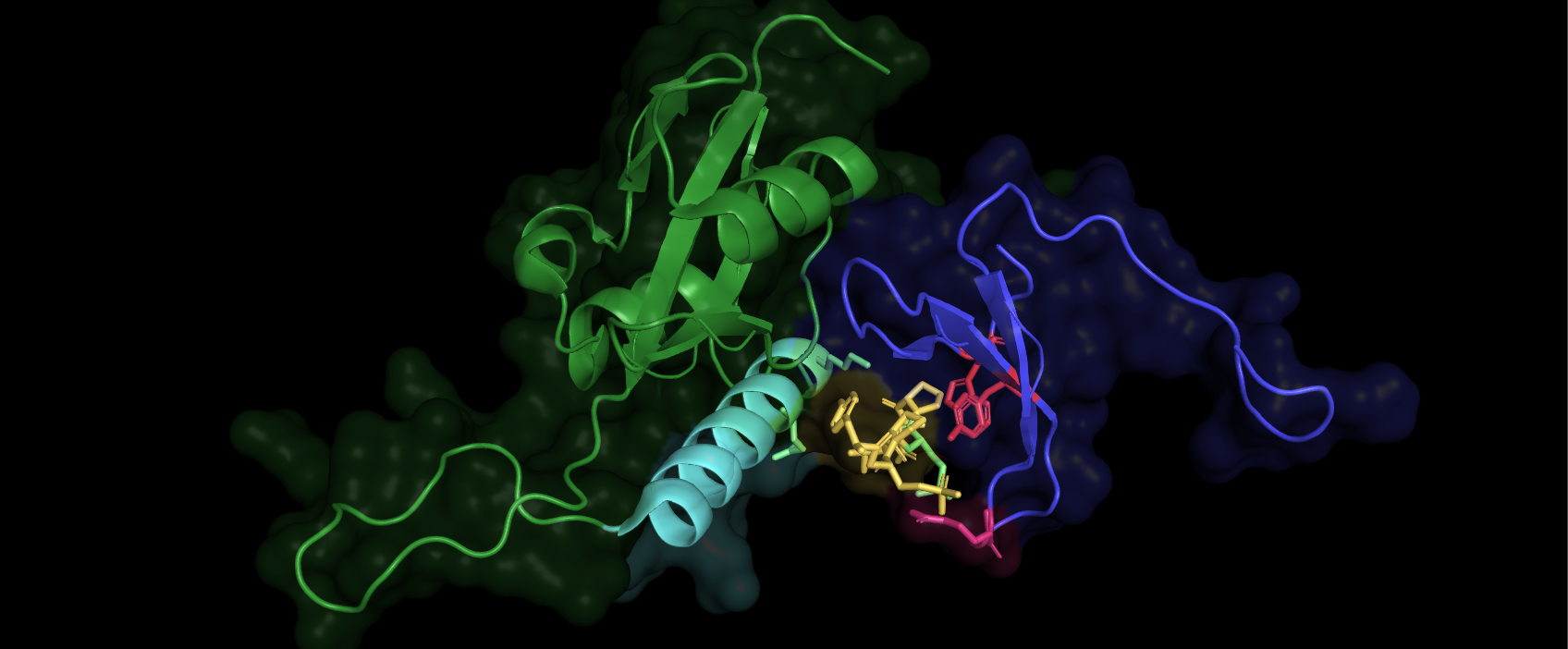
We can see clearly that the lower range of the color scale, blue, is represented pocket close to tyr 26 trp 37 and arg 17, which holds if down rigidly. The red part of the peptide has high disorder and are bound much weaker, it can be seen to stick out and be unsupported by the binding pocket.
To get a better understanding of the domains WW / PPiase / linker, I would select the WW region and save it like we did with the other parts. The ww domain is N-terminal in Pina1 so mark residues 1-41 and name them WW domain.
Now if you hide the part we named Pin1, we and show the part called ww-domain as ribbons, we can see clearly how it is positioned in regard to the peptide that has bound to the WW domain. If you want to, you can chose show surface, color it e.g purple and the hit the command: Set transparency, 0.7, 0 = no transparency 1 = 100% transparency, you can play with the setting to get a good view. Mark the region 42-End of sequence or residue 166, name it PPiase + Linker, show surface, green, and show the surface on the peptide you named, color it blue. This is what we can so now:
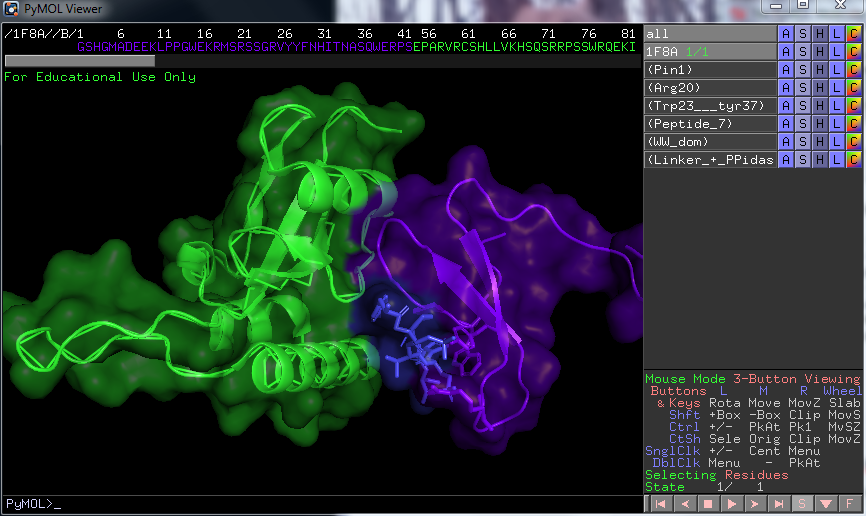
The surfaces correspond well to the electron cloud of the amino acids / protein, and determines the range of interaction. The electron cloud is also the structure we directly map from crystallography data, using bragg's law. We will then embed the atoms in the cloud via the best fit. The result is seen above. This crystal structure is frozen and compact so we must use an NMR structure taken in solution, with no protein bound to it, to see how the structure looks in vivo(in our cells). We can see above that an alpha helix sits close the the peptide bound to the ww domain(containing arg17, trp/try 26/37) and might be important for the binding. We will also be able to conclude if the protein´s linker is involved in forming the active PPiase domain.
As you might suspect, a lot of information about the peptide binding to the WW domain on an atom scale, can already be draw:
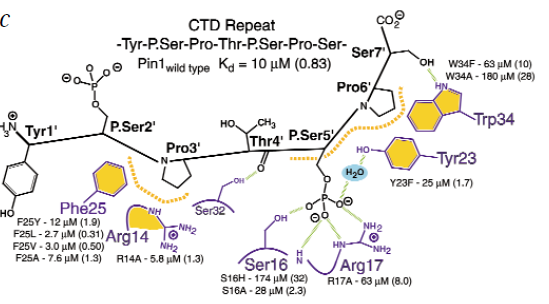
This is very good information indeed, and clearly shows how Arg17 is the main hydrogen bond donor, from both its side chain nitrogen as well as the backbone amide group(nitrogen and hydrogen group). We can see that tyr 23 (27 in our sequence due to 4 extra residuals on the N-terminus) also is involved in orienting a water molecule via hydrogen bonding and the water in it´s turn, stabilize the phosphate head on the 3rd oxygen. Zoom in and click the interesting residuals, chose lable residual, and you can see this:
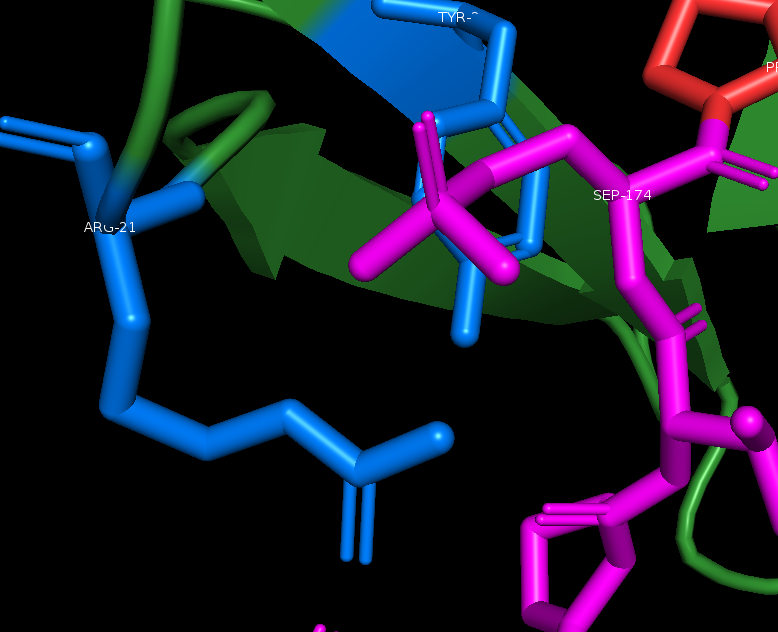
Bleow in the next image I have labeled typ and trp 27 & 38, we can see clearly that tyr 23 and trp 34(27 & 38 in our sequence) is working as a clamp, and that a longer side chain would not fit here. This makes proline an optimal fit, the ring Proline has, generated by binding it´s sidechain back to the backbone amide group, interacts with the tyrosine, as well as fits perfectly in the thigh space.
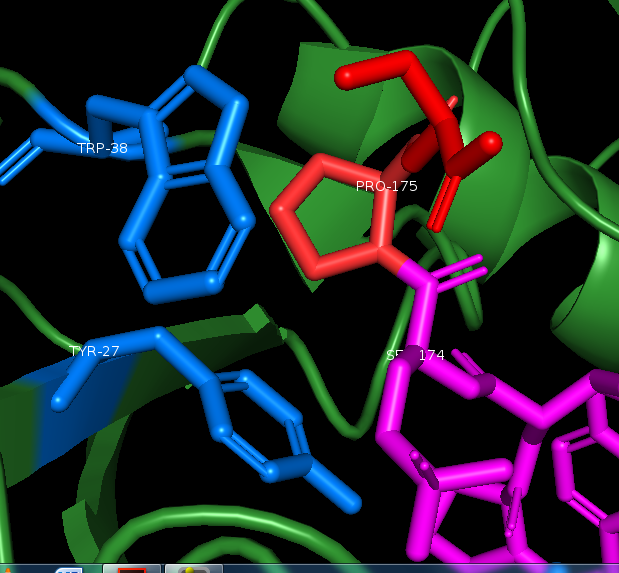
This also illustrates well, how specific the domain is for phosphate. Trying to bind Pin1 to the same peptide with no phosphate on the serine, will cause total loss of binding. This points to how tightly regulated its activity is.
Prior to going on with the NMR structure, this should be known:
Mutation of Arg 17 causes losses of 6 fold affinity, so it is very important for overcoming the energy barrier of binding.
We also can show residuals 71 / 72(67 & 68 in a non extended sequence) to so that they are involved in forming the active PPiase domain. This means that the Linker where these residuals sits, must pull towards the PPiase site to activate it, pointing to cooperativity via an allosteric mechanism to enable specific and active binding.[2]
PPiase - Catalytic domain and the Linker - Making Allosteric cooperative effects possible. NMR
Now we will fetch a now file, just type" fetch 1NMV" in a new session, and the NMR structure will load.
Again start with pressing the S on the bottom to see the full sequence and the the S on the entry in the list, named All, and hide everything. Now we have a black screen, mark all the amino acids in the sequence on top and rename the new list entry called sele. Call it Pin1 and press the S next to it, choose show as ribbons and the protein will appear. We can see a totally different conformation here, while in solution and not bound to a protein. The WW domain 1-41 can be market and colored blue, just as always, rename it to WW, to easily see where it sits when in solution.
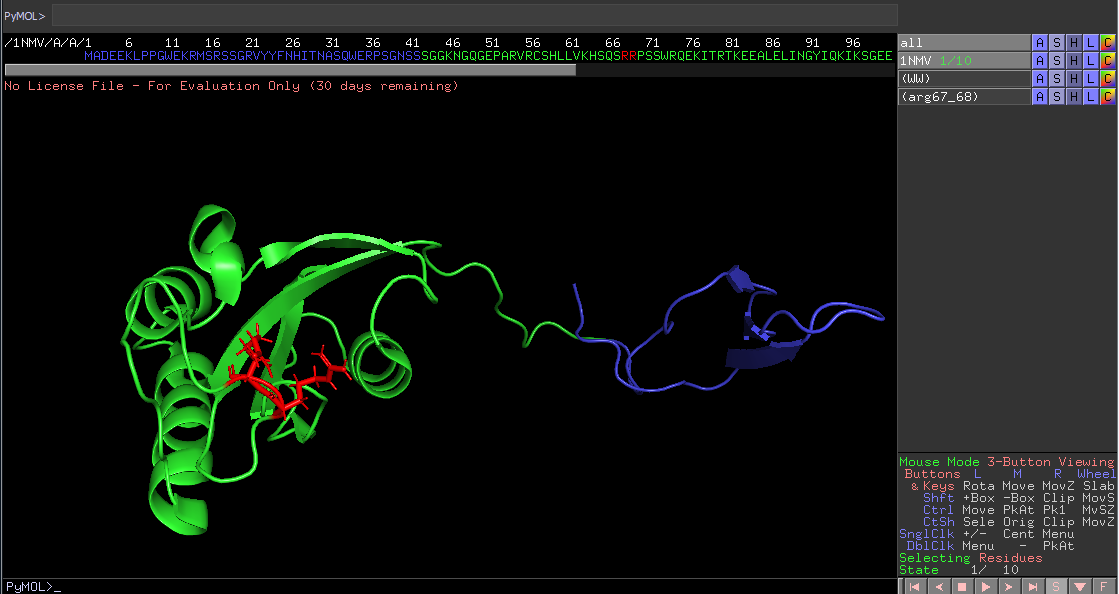
It is open and flexible as we suspected, and the linker residuals 68/69 both arginine, seen in red, indeed will move and help form the PPiase pocket by pulling the protein to its more compact state. This is not an single even, many chemical interactions reverberate through the linker, but the trigger is binding to Ser.P-prolin motifs. This structure resulting will not be as compact as the crystal structure would suggest, due to the crystal form and low temperature. It is only bound to 7 residuals, which will limit the understanding since in vivo a big protein would bind and limit Pin1´s possibility to become so compact. This also proves that the WW pocket is not complete without being altered, the alpha helix we saw in the x-ray pictures, is now sitting to the bottom left(the biggest helix) and must be moved quite a bit to support the pocket structure of the WW domain, another event pushing for the compact state. This movement of helix and pulls on the linker. Finally the PPiase domain will be open and ready to its designated protein in vivo.
Expressing only the PPiase region of the protein will cause total loss of affinity, but still have some catalytic effect, which points to the fact that WW domains are specificity filters.[3]

You can off course fetch how many structures you want, here is 1NMV and 1F8A side by side and oriented so we can see that the WW part, smaller region, with just three bets sheets, will be oriented on top of the PPiase region, and in vicinity of the largest alpha helix. The blue, folded state could be rotated 180 degrees upside down, to see better how it fits, I ll leave that to you. Just zoom and rotate with the mouse, maneuvering is easy, but should you end up in trouble feel free to leave questions in the comment section.
--
This is what happens if you chose movie and rotate the protein around the Y-axis 180 degrees over 18 sec, from the menu, then export movie and upload it at Dtube, or if it wont work, another site.
I tried @Dtube first, but it wont take the file :( But I just noticed it breaks down after 9 sec at least partially, and is 18 sec long so that was for the best perhaps, but its worth a look :) It could have to due with the missing sound format. Same settings as the first image is used, just played with colors and regions a bit.
References:
1: http://www.genecards.org/cgi-bin/carddisp.pl?gene=PIN1
2: Structural basis for phosphoserine-proline recognition by group IV WW domains Mark A. Verdecia, Marianne E. Bowman, Kun Ping Lu, Tony Hunter and Joseph P. Noel1Structural Biology Laboratory and Department of Biology,
University of California. https://www.nature.com/articles/nsb0800_639
3:Structural Analysis of the Mitotic Regulator hPin1 in Solution: INSIGHTS INTO DOMAIN ARCHITECTURE AND SUBSTRATE BINDING.
Received for publication, January 22, 2003, and in revised form, April 22, 2003 Published, JBC Papers in Press, April 29, 2003, DOI 10.1074/jbc.M300721200
And off course PyMOL by Schrödinger: Thanks for the great software!
All structures are available at https://www.rcsb.org/ there are 140,000 in total so don't be afraid to view through them all to fast :)
Thanks for checking my work out, It is much appreciated.

Thank you for the link to PyMol, what a great tool. I'm taking an organic chem course and it will give me a visual of the protein changes.
I appreciate your support, thank you. Upvoted.
Thanks for checking it out :) Likewise!
I'm hoping it will help me understand how resonance works.
Nice one @clausewitz
I have worked on this platform once in my masters, now I just to use it again for some short peptides. There is something you might help.
Instead of taking any PDB ID or a protein of higher size (around 5-20 KDa), I want to make it for a tri or pentapeptide with modifications at both the terminals.
Is it possible to make one?
And, these peptides will be further self-assemble to give mostly beta-sheets. So I want to make a demo using pymol.
Hey, yeah Pymol have a builder tool which is easy to use for peptides and modifications, but making something self assemble is another question :).
Just click builder, residuals and pick the amino acid you want, 1 by 1 and you have the designed peptide in pymol.
Here is the tritide DQV in pink, and sticks, The white ball thing tells you where next residual will sit.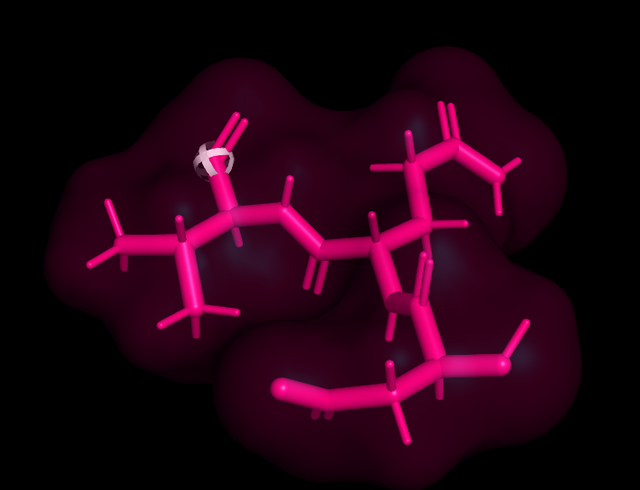
It might be hard to do a self assembly, or I have never animated such interactions in anyway.
I just tried to import many peptides, that´s no problem but to make frames of value is hard, you could move them 1 by 1 and make frames, and beta sheet can be formed lastly.
But I think pymol is hard to use for this purpose!
I ll come back if i find something.
Thanks @clausewitz
Addiion of amino acid I can do but unable to add any other organic group to the the terminals. Is it possible?
If I am working with a pentapeptide and want to make a model of its secondary structure. A single molecule is not going to form the whole structure only after assembling themselve it will form structure. If I know the typeof interaction happening in it, can I make one from pymol also which dipicts the whole phenomenon.
Thanks
Hey! Just build the peptid, click 1 atom, then now use the alternative under build called framgnet, click what you like to add.
Here is two amino acids imported on each other, both with a bromine group, and lastly a cyclo pentante on the lower residual, closing both of the Amino acids in:) just for the fun look of it :)
Cyclopenate in Pink + Bromine in red.
It worked..... thanks
I'll see for the assembling part
Thanks once again
Anytime :)
Yeah that part is harder, and after talking to some colleges we would recommend a graphical designer, not engineers in biochemistry hehe
Best of luck!
Great post. I shall definitely take a look at this software!
Yeah it is sweet if you want make molecules or binding zones visible, you can zoom in very far and watch single atom positioning etc
I am trying to download it as we speak, and look forward to doing some modeling on my favorite proteins. Cheers!
The name "Pin1" has (unfortunately) also been used for a plant protein, and I think this may be a potential source of confusion in the literature. Pin1 in plants is involved in polar auxin (indole acetic acid) transport: Xi et al. Pin1At regulates PIN1 polar localization and root gravitropism. Nature Communications 7: 10430 (2016). I believe that this plant "Pin1" has a completely different structure, but please correct me if I am wrong. I look forward to following your posts in the future.
Yeah this seems solid, its about Pin1at, and the PIN in this article refers to: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2812941/ an abbreviation for something else.
You are right they are very different Pin1 vs PIN which comes in versions 1-7 :)
Thanks for the contribution I appreciate it, so no confusion strikes, should someone stumble on another PIN1, the auxin transport proteins, PIN 1,2,3,4,5,6 or 7 or pin1at in palnts for that matter.
The structures I used, are expressed in models systems, with human gene sequence as template.
The article only mentioned Pin1 here, so I get the possible confusion! PPias-type :)
The opening statement explains the PIN abbreviation. The only link from PIN to Pin1 is that a Pin1 similar protein called Pin1at activates or regulates PIN ergo PIN-FORMED :)
Pin1at, the orthologue in the studie. Lacks WW domain here, but are similar!
Thanks again, good information to have here :)
Congratulations! This post has been upvoted from the communal account, @minnowsupport, by Clausewitz from the Minnow Support Project. It's a witness project run by aggroed, ausbitbank, teamsteem, theprophet0, someguy123, neoxian, followbtcnews, and netuoso. The goal is to help Steemit grow by supporting Minnows. Please find us at the Peace, Abundance, and Liberty Network (PALnet) Discord Channel. It's a completely public and open space to all members of the Steemit community who voluntarily choose to be there.
If you would like to delegate to the Minnow Support Project you can do so by clicking on the following links: 50SP, 100SP, 250SP, 500SP, 1000SP, 5000SP.
Be sure to leave at least 50SP undelegated on your account.