Otocephaly: a case postnatal diagnosed
Otocephaly is characterized by agenesis or severe hypogenesis of the mandible or agnathia, synotia (the external ears horizontally placed and/or fused), microstomia (“small mouth”), aglossia (congenital absence of the tongue). This anomaly is often associated with other malformations as; Holoprosencephaly, anencephaly, congenital heart
disease, tracheoesophageal.
The etiology is unknown. The pathogenetic mechanism seems to be related to a complete failure of the mandibular development, possibly associated with inhibited or arrested migration of neural crest cells. Three-dimensional ultrasound is often the only way to obtain an overall idea of the anomaly. The virtually all views of the fetal face are abnormal, due to the complete distortion of the facial anatomy. Otocephaly has been always lethal, so when detected, an accurate counseling of pregnancy should be offered at parents with also the possibility to offer termination of pregnancy. We report the case of a fetus, who was born on the 30th week of pregnancy, and was diagnosed with otocephaly, after birth.
A 21-year-old G2P1A0 patient presents at 30 weeks gestation for labor with uterine contractions, and is found to be in active labor with the fetus in breech presentation. Clinical assessment of the maternal pelvis is determined to be adequate for a fetus of this estimated weight.
The cervix is 10 cm dilated, 100% effaced and the fetal pelvic are at -3 station. Ultrasound examination reveals of polyhydramnios, was reconfirmed afterwards with rupture of
membrane he had approximately 5 L amniotic
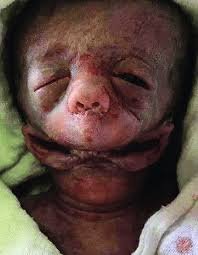
fluid. The patient had no medical and family history of any congenital anomalies, and likewise had no antenatal history of infection/radiation or toxin exposure. She has had a non-good prenatal care, including lack of one anatomy scans in the second trimester. Her prior infant, weighing approximately 3500 g, was delivered vaginally without complications. He was completely healthy. After 30 minutes, the patient with vaginal delivery, with manual assistance, according to Bracht maneuver, gave birth to a male baby with body weight 900 g and Apgar score 4/5. The macroscopic placenta was normal, and the umbilical cord with central insertion and that had three vessels. A detailed examination showed multiple anomalies of face as; Synotia (the external ears horizontally placed and/or fused), Agnathia (agenesis or severe hypogenesis of the mandible), Microstomia (“small mouth”), Aglossia (congenital absence of the tongue). Based on these components was diagnosed as an otocephaly. Six hours after delivering the baby, he dies.
Otocephaly is characterized by agenesis or severe hypogenesis of the mandible (agnathia). The temporal bones are juxtaposed and the external ears are horizontally placed and/or fused. Different degrees of severity have been described (synotia, agnathia, microstomia, aglossia etc.). This anomaly is often associated with other malformations as; holoprosencephaly, anencephaly, congenital heart disease, tracheoesophageal fistula etc. The etiology is unknown. Some report’s emphasis to the role of autosomal recessive inheritance and drugs as possible causes
(1-4)
. Otocephaly is an anomaly that has not been reported to be associated with trisomy 13
(5,6)
. The incidence of the otocephaly is around 1 in 70,000 births
(2)
. The pathogenetic mechanism seems to be related to a complete failure of the mandibular development, possibly associated with inhibited or arrested migration of neural crest cells. Three-dimensional ultrasound is often the only way to obtain an overall idea of the anomaly. Otocephaly ultrasound diagnosis can be made on the midsagittal view of the profile, where to all views of the fetal face are abnormal, due to the complete distortion of the facial anatomy. The fetal face represents an extremely irregular region to explore with ultrasound. This is why ultrasound examination of the fetal face has always posed great difficulty during the examination. We have some scanning planes, classified as axial, coronal, sagittal, and oblique, and the anatomic structures assessable in each of these views. In an axial view are usually seen; tongue, pharynx, Mandible, inferior alveolar ridge, mandibular bone etc. In a sagittal view are usually seen; facial profile (midline sagittal) and ear—parasagittal (lateral). The final scanning planes to complete the evaluation of the splanchnocranium are as follows:
- The coronal view of the face and palate;
- The oblique view of the upper lip;
- The coronal/oblique view of the palate.
Scanning for fetal anomaly is usually performed between 18 and 22 weeks’ gestation. Otocephaly has been always lethal, so when detected, an accurate counseling of pregnancy should be offered at parents with also the possibility to offer termination of pregnancy. In such cases, postnatal treatment is not possible. Our case was a malformation of order that consisted of synotia, agnathia, microstomia, aglossia. A case that after six hours end to death. In the pathogenic aspect , during the 4th weeks gestation, the mesoderm lateral plate of the ventral foregut region becomes segmented to form a series of five distinct bilateral mesenchyme swellings called the pharyngeal (branchial) arches. Ventrally migrating neural crest cells interact with lateral extensions of the pharyngeal endoderm, surround the six aortic arch arteries, and initiate pharyngeal arch development. The initial mesodermal core of each arch is augmented by neural crest tissue that surrounds the mesodermal core. The mesoderm will give rise to muscle myoblasts while the neural crest cells give rise to skeletal and connective tissues.
Otocephaly: a case postnatal diagnosed Astrit Gashi et al.
The pharyngeal arches are separated by pharyngeal grooves on the external aspect of the embryo, which correspond internally with five outpouchings of the elongated pharynx of the foregut, known as the five pharyngeal pouches. Although derivatives of five or even six arches are described, only four arches appear externally. The first pharyngeal arch is the precursor of both the maxillary and mandibular jaws and appropriately bounds the lateral aspects of the stomodeum. Meckel’s cartilage arises at the 41st to 45th days post conception, provides a template for subsequent development of the mandible. Persisting portions of Meckel’s cartilage form the basis of major portions of two ear ossicles. Deficient development of the pharyngeal arches results in syndromes that are identified according to the arch involved. The syndromes become rarer as the number of the arch increases. Severe first-arch anomalies are: Agnathia, Synotia, Microstomia. Less severe are: Treacher Collins syndrome, Pierre Robin syndrome etc.
Congratulations @cholunga! You received a personal award!
You can view your badges on your Steem Board and compare to others on the Steem Ranking
Vote for @Steemitboard as a witness to get one more award and increased upvotes!