Mito-Medizin: Trend oder Tatsache? Part II – Von den Mitos zum zellulären Gezwitscher.
In diesem Beitrag wollen wir uns näher mit den Konsequenzen mitochondrialer Krankheiten beschäftigen. Bei der Recherche zum Thema konnte ich feststellen, dass es in der Tat viele Ansichten gibt, wann man von einer mitochondrialen Dysfunktion überhaupt sprechen kann. Die einen sehen eine mitochondriale Dysfunktion auf Grundlage von Mutationen in mit Mitochondrien-assoziierten Genen an, welche in ihrer Endkonsequenz die Funktionsfähigkeit der Mitochondrien mindern. Andere fassen die Problematik etwas weiter (so auch ich) und würden eher alle Beeinträchtigungen der Zelle, welche letztlich die Leistung der Mitochondrien herabsetzen miteinbeziehen.
Mitochondriale Dysfunktion sind, sofern sie vererbt wurden, mit erheblichen Problemen für den Menschen verbunden [1, 2]. Das bedeutet, dass die meisten Menschen entweder überhaupt nicht lebensfähig sind (also früh in der Entwicklung oder spätestens im Kindesalter versterben) oder spätestens als Erwachsene erhebliche gesundheitliche Probleme haben. Beispiele für solche Probleme sind unter anderem Taubheit, Blindheit, Herzprobleme, Diabetes, Leberschäden, Muskelschwächen und allgemeine Erschöpfung bis hin zu neurologischen Problemen wie Depressionen und Demenz [1-6].
Da viele der Erkrankungen multifaktorielle sind, ist es allerdings sehr schwierig einzuschätzen woher die Probleme hauptsächlich stammen. Die Kenntnis über die Ursache einer Erkrankung ist aber absolut entscheidend, um eine konkrete therapeutische Konsequenz abzuleiten zu können. Und Mitochondrien sind prädestiniert für solche multifaktoriellen Probleme, denn wie ihr aus dem ersten Teil wisst, sind diese eifrigen Gefährten an eigentlich allem was in der Zelle und letztlich im Körper passiert irgendwie beteiligt.
Die mitochondrialen Dysfunktionen stellen somit eine enorme Herausforderung für Ärzte, Wissenschaftler und letztlich die Betroffenen dar.
Gegenwärtige Schätzungen kommen zu dem Schluss, dass etwa eine von 2000 Personen an einer mitochondrialen Dysfunktion leidet [1]. Diese Schätzungen berufen sich allerdings vorrangig auf Personen, welche auch eine Mutation in einem von 1300 Genen, welche im Zellkern oder einem von 37 Genen, welche in den Mitochondrien selbst kodiert sind, zeigen [1, 2]. Nicht berücksichtigt werden jedoch oft die Probleme im zellulären Gefüge, die ebenfalls die mitochondriale Leistungsfähigkeit beeinträchtigen [6]. Zu diesen gehören beispielsweise Probleme in den zuführenden Pathways, in Recycling-Mechanismen oder die Exposition zu toxischen Substanzen (wie Antibiotika oder Schwermetalle), welche die mitochondriale Leistungsfähigkeit beeinträchtigen können [7].
Der einschlägigen Literatur nach zu urteilen ist es gegenwärtig überhaupt sehr schwierig angemessene Therapien vorzuschlagen, denn z.B. einen Gendefekt ist momentan noch nicht korrigierbar [1, 8]. Solche Interventionen wurden zwar schon oft erprobt, jedoch ist eine flächendeckende sichere Anwendung noch in weiter Ferne [8]. Darüberhinaus wird es umso schwerer solche Probleme zu beheben je mehr Gene betroffen sind.
Vielleicht sollten wir uns aber erst einmal einen Überblick über mögliche Szenarien verschaffen, um zu verstehen woher die Probleme stammen und welche Möglichkeiten es geben könnte diese vielleicht zu vermeiden oder im Ernstfall mild zu behandeln.
Der gegenwärtigen wissenschaftlichen Literatur nach können primär fünf Hauptursachen für mitochondriale Dysfunktionen definiert werden:
- Mutationen der mitochondrialen DNA, welche mit der Herkunft verbunden sind und schon viele Jahrhunderte/Jahrtausende in einer bestimmten Volksgruppe vorhanden sind.
- Mutationen der mitochondrialen DNA, welche erst „kürzlich“ eingebracht wurden.
- Mutationen in der mitochondrialen DNA, welche man im Laufe des Lebens anhäuft.
- Mutationen von Genen in der Kern-DNA, die für mitochondriale Genprodukte kodieren.
Und schließlich: - Mutationen, die den Crosstalk zwischen Mitochondrien und Zellkern beeinflussen.
Ich würde jedoch noch einen Schritt weitergehen und Punkt 6. definieren als: - Störungen des Crosstalks zwischen Mitochondrien und anderen Zellorganellen, wie dem Endoplasmatischem Retikulum (ER).
Außerdem ist meiner Meinung nach noch Punkt 7. wichtig, nämlich: - Störungen im Metabolismus und in Signalling Pathways, die eine optimale Mitochondrienfunktion torpedieren.
Ich würde vorschlagen alle Probleme einzeln zu beschreiben und zu überprüfen inwiefern diese vermieden, gelindert oder ganz geheilt werden können.
Beginnen wir mit Punkt 1.: Mutationen, welche charakteristisch für bestimmte Volksgruppen sind.
Diese Mutationen haben sich innerhalb von Generationen im mitochondrialen Genom von Menschen eingenistet und konnten sich deshalb dort halten, weil sie u.a. einen Vorteil, keinesfalls aber einen dramatischen Nachteil zum Überleben darstellten [2]. Diese Ansammlungen von genetischen Besonderheiten in geographisch klar definierten Bevölkerungsgruppen, die sich von einem gemeinsamen Vorfahren ableiten, werden als Haplotypen bezeichnet [2, 4, 9].
Abb.1 Haplotypen dieser Erde. Beachtet, dass es haufenweise verschiedene Haplotypen gibt. Das Bild soll den Text lediglich etwas auflockern. Wenn ihr mehr über eure potentielle „Haplotypenzugehörigkeit“ erfahren wollt, dann schaut einfach mal hier vorbei. Made by Chapper - unrestricted use allowed
Sie waren oft Adaptationen an das vorherrschende Klima, die verfügbaren Nahrungsquellen und vieles mehr [2]. Einst beeinflussten die Umweltbedingungen sehr stark das Leben der Menschen. In Zeiten von Überfluss, guter Infrastruktur und hervorragender hygienischer wie medizinischer Versorgung, ist dies allerdings kein Thema mehr. Eher ist das Gegenteil der Fall und so kann sich beispielsweise kalorienreiche Nahrung für manche Menschen als sehr nachteilig herausstellen [4]. Doch auch Langlebigkeit wird unter anderem mit Haplotypen in Verbindung gebracht [2].
Wer mehr über Haplotypen erfahren möchte, der kann auf der Ancient mtDNA database vorbeischauen und sich vielleicht selbst sequenzieren lassen.
Werde ich bestimmt auch bald mal tun!
Behandelbar sind solche Mutationen ohne genetische Interventionen wahrscheinlich nicht. Der Grund ist einfach, dass diese Mitochondrien einer Selektion unterlagen und sich dabei gegen andere Mitochondrien durchsetzen konnten. Die Mitochondrien, welche ihr von eurer Mutter erhalten habt (Mitochondrien werden immer mütterlicherseits vererbt), sind demnach bzgl. diese Haplotypen reinerbig. Obwohl es mittlerweile Zweifel darin gibt [10].
Gleiches gilt prinzipiell auch für Punkt 2.: Mutationen, welche sich in vergleichsweise kurzen Zeiträumen ergeben haben.
Auch hier sind (der Literatur nach) genetische Eingriffe notwendig [1]. Die Kategorie umfasst, meiner Meinung nach, die schwerwiegendsten Probleme. Zu den „milderen“ Probleme dieser Art gehören z.B. Diabetes Mellitus Typ II, geringe Belastbarkeit bei sportlichen Anstrengungen oder Taubheit [2]. Alle anderen wirken sich in extremer Art und Weise auf das Leben der betroffenen aus. Beispiele hierfür sind u.a. MELAS. MELAS ist eine Krankheit welche sich u.a. durch Mitochondrien-assoziierte Hirnschäden (Enzephalopathien), starke Ansäuerung des Blutes (Laktische Azidose), sowie durch schlaganfallartige Episoden (Stroke) äußert. Verantwortlich ist hier, allem Anschein nach, oft nur eine einzige Mutation in einem einzigen mitochondrialen Gen für eine tRNA, welche die Aminosäure Leucin zu den mitochondrialen Ribosomen transportiert (Abb.2) [1, 2].
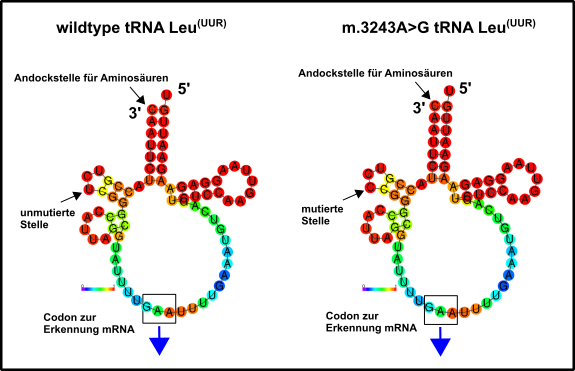
Abb.2 Mitochondriale tRNA für die Aminosäure Leucin. Links die Wildtyp-Variante, rechts die Variante, welche bei der Erkrankung MELAS mutiert sein kann. Die ursprüngliche mtDNA-Sequenz, welche diese tRNA kodiert fand ich hier. Danach habe ich zunächst die komplementäre Sequenz erstellt, weil die Transkription immer antiparallele Produkte hervorbringt. Dies gelang mir mit diesem Tool hier. Schließlich wurde das Produkt in RNA umgeschrieben, denn RNA trägt beispielsweise statt einem Thymin (T) ein Uracil (U). Dies umzuschreiben ist aber dank diesem Werkzeug hier kein Problem. Letztlich wurde mit dem RNAFold WebServer ein mögliches Modell der beiden tRNAs erstellt. Dargestellt ist die Tendenz zur Knüpfung von Bindungen. Aber auch die Entropieverteilung zeigte mir keinerlei gravierende Unterschiede an. Entweder ich muss dies künftig noch etwas ausgefeilter machen oder der Nachteil dieses Basenausstauschs ergibt sich durch direkte Wechselwirkungen, z.B. mit dem Ribosom, welche nach Mutation nicht mehr oder nur noch eingeschränkt gewährleistet sind. Made by Chapper - unrestricted use allowed
Jedem ist hoffentlich klar, dass Leucin eine wichtige Aminosäure ist, deren Abwesenheit einen korrekten Zusammenbau der mitochondrialen Proteine gar nicht oder nur unzureichend ermöglicht [11, 12]. Diese kleine Mutation hat demnach erhebliche Auswirkungen. Wenn ihr mich fragt sollten sich solche Erkrankungen aber in naher Zukunft sehr gut behandeln lassen. Ähnliches ist auch im Falle der Erkrankung MEERF gegeben, die noch viel zahlreichere Symptome aufweist, ebenfalls aber häufig auf eine einzelne Mutation in einer mitochondrialen tRNA reduziert werden kann [1]. Oberflächlich betrachtet scheint es, als ob Mutationen in mitochondrialen tRNAs den Löwenanteil der mitochondrialen Dysfunktionen in Kategorie 2 ausmachen. Die Menschen mit diesen Problemen sind häufig schon als Kinder betroffen, die Probleme verschlimmern sich, wie beim Charcot–Marie–Tooth-Syndrom, aber bis ins Erwachsenenalter immer mehr und mehr [1]. Wer Leute kennt, die an sowas leiden weiß, dass es oft ein mühseliger Kampf um etwas Lebensqualität ist. Nicht zu fassen, dass eine einzige falsche DNA-Base dafür verantwortlich sein soll. Einer meiner ehemaligen Diplomanden leidet auch an sowas, aber er ist ein Kämpfer. Respekt! Und Respekt natürlich auch gegenüber all den fleißigen Genetikern, die hoffentlich bald eine Möglichkeit gefunden haben, um dies zu korrigieren.
Dennoch frage ich mich ob die Annahme, dass erst genetische Eingriffe eine Linderung bringen könnten korrekt ist, denn immerhin verschlimmern sich die Probleme zum Erwachsenenalter hin. Die Mitochondrienerkrankung LHON beispielsweise führt erst im zweiten oder dritten Lebensabschnitt zu allmählicher Erblindung [3]. Irgendwas muss demnach im Zeitraum bis dahin im Körper passieren, was später diese Auswirkungen zeigt. Wir kommen später nochmal darauf zurück.
Kommen wir erstmal zu Kategorie 3.: Mitokrankheiten, die sich im Laufe des Lebens ansammeln. Sogenannte somatische Mutationen.
So hier wird es jetzt ziemlich spannend wie ich finde und hier ist es auch höchste Zeit mein Inkscape wieder richtig startklar zu machen. Prinzipiell diskutieren wir diese Problematik schon die ganze Zeit während ich diesen Blog hier mache und jedem Stammleser sollte klar sein was jetzt kommt. Richtig: Euer Life-Style. Wenn die Leute aus den ersten beiden Kategorien nun wirklich nix für ihr Leid können, so kommen wir jetzt in den Bereich des (potentiell) selbstverantworteten Leids. Lasst es mich kurz illustrieren.
(Ich beziehe mich in dieser Kategorie primär auf meine bereits angefertigten Post über Autophagie, sowie meine Reihe über gesundes Leben, eine Zusammenstellung findet ihr hier).
Dies sind eure Mitochondrien, wenn ihr jung, frisch und knackig seit:
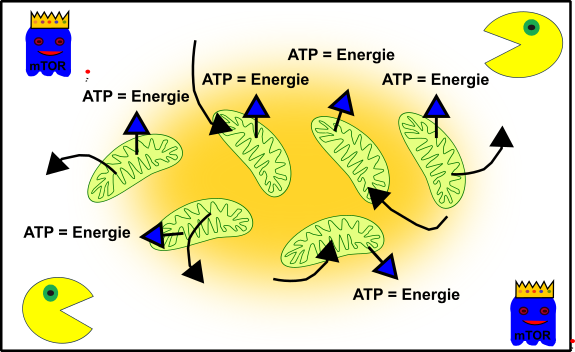
Abbildung 3: Vereinfachte Darstellung eures zellulären Gefüges in jungen Jahren. Eure Mitochondrien (grün), Autophagie (Packman) und fördernde Pathways (Gespenst in blau mit Krone). Made by Chapper - unrestricted use allowed
Eure Mitos produzieren eifrig Energie damit ihr die ganze Zeit unermüdlich rumrennen könnt‘. mTOR (das blaue Gespenst) lässt euren Körper schön wachsen und gedeihen. Und euer zellulärer Packman (Autophagie) passt auf, dass alles schön sauber und ordentlich ist. Die perfekte Symbiose, wenn ihr wollt. (Kleine Anmerkung an meine Neuleser, falls euch das gerade etwas bekloppt vorkommt, dann checkt bitte meine anderen Posts zum Thema, Danke!).
Nun aber werdet ihr älter und alles gerät etwas in Schieflage. Zum Beispiel habt ihr vielleicht etwas mehr Kummer, esst die ganze Zeit bei McDonalds, fangt an zu Rauchen und zu saufen oder Schlimmeres. Nun ist das Gefüge nicht mehr ganz so prickeln wie einst:
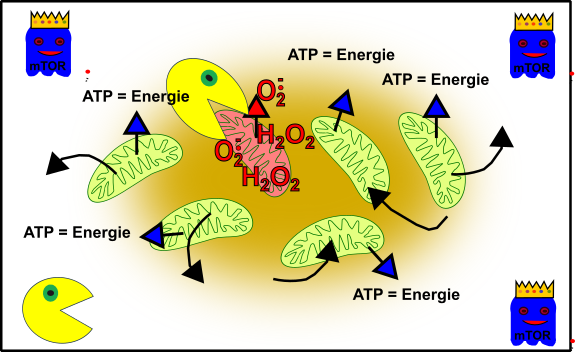
Abbildung 4: Vereinfachte Darstellung eures zellulären Gefüges im Verlauf eurer Adoleszenz. Eure Mitochondrien (grün), durch oxidativen Stress beschädigte Mitochondrien (rötlich), Autophagie (Packman) und fördernde Pathways (Gespenst in blau mit Krone). Made by Chapper - unrestricted use allowed
Der erhöhte Stress in euren Zellen hat in einigen Mitochondrien zu erheblichen Schäden geführt. Oxidativer Stress (z.B. H2O2) hat diese Mitochondrien außer Gefecht gesetzt. Aber keine Panik! Euer zellulärer Packman (Autophagie) bereinigt die Sache und weiter geht’s.
Jeder weiß, dass es stets immer schwieriger wird im Leben und auch eure Zellen haben mit den Jahren mehr und mehr zu kämpfen. Nicht nur, dass euer zellulärer Packman zunehmend fauler wird [13], nein auch mTOR zeigt mehr und mehr sein wahres Gesicht und verhindert nicht nur, dass Zellen, die am Ende sind endlich sterben [14], im Gegenteil, mTOR arbeitet aktiv gegen unseren lieben Packman [15]:
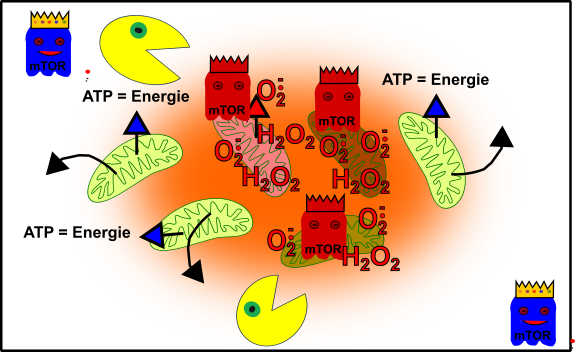
Abbildung 5: Vereinfachte Darstellung eures zellulären Gefüges im Erwachsenenalter. Eure Mitochondrien (grün), durch oxidativen Stress beschädigte Mitochondrien (rötlich), Autophagie (Packman), fördernde Pathways (Gespenst in blau mit Krone) und aus dem Gelichgewicht geratene fördernde Pathways (rote Gespenster mit Krone). Made by Chapper - unrestricted use allowed
Nun beginnen die Probleme. Du bist schwach, ausgebrannt, überall Schwierigkeiten. Keine Leistung, keine Kraft, ständig hast du irgendwas. Du denkst: „Diese oder jene Pille wird schon helfen!“ „Ein kleiner Absacker hat noch niemandem geschadet!“ „Was kann an ein paar aufmunternden Süßigkeiten schon verkehrt sein, wo ich doch heute so niedergeschlagen bin?“ In Wahrheit verschlimmern all diese Interventionen aber alles noch viel mehr, denn mTOR hat nur darauf gewartet, um euren zellulären Packman noch mehr zu unterdrücken. Der Terror nimmt seinen Lauf. Die Mitochondrien, die einen totalen Knacks haben, werden nun nicht einfach beseitig, vielmehr vermehren sie sich auch noch [4]. Und so habt ihr am Ende die Mutationen, welche schwerkranke Menschen in ihren Mitochondrien von Anfang an tragen (+X), ebenfalls in euerem Bestand.
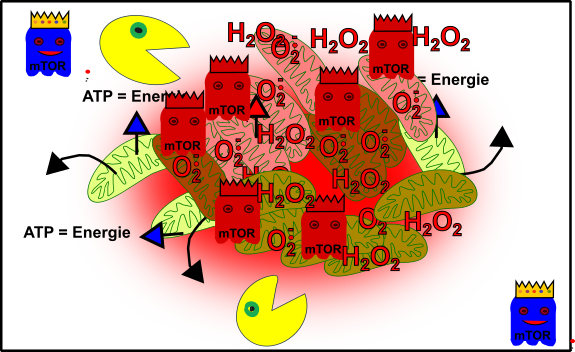
Abbildung 6: Vereinfachte Darstellung: Euer zelluläres Gefüge ist im Eimer! Eure Mitochondrien (grün), durch oxidativen Stress beschädigte Mitochondrien (rötlich), Autophagie (Packman), fördernde Pathways (Gespenst in blau mit Krone) und aus dem Gleichgewicht geratene fördernde Pathways (rote Gespenster mit Krone). Made by Chapper - unrestricted use allowed
Herzlichen Glückwunsch! Du hast erfolgreich deine Gesundheit ruiniert!
Und nun? Wie geht’s weiter? Tja, ich kann nicht alles eine Million Mal wiederkäuen, check meine anderen Posts zu vergleichbaren Themen und du wirst eine Antwort finden.
Punkt 4. Ist wieder weniger selbstverschuldet: Mutationen in der Kern-DNA.
Dies ist ein besonders spannender Aspekt wie ich finde, denn wie ihr bereits aus dem ersten Teil dieser Serie wisst, haben die Mitochondrien nach ihrer „Übernahme“ (Endosymbiose) zahlreiche Gene in den Kern ausgelagert [1, 2, 4, 16]. Sage und Schreibe 1300 Stück(!). Nur 37 Gene sind in den Mitochondrien selbst verblieben (wenn auch die Wichtigsten). Nichtdestotrotz sind diese 1300 Gene nicht minder notwendig [2]. Die Komplexe der Atmungskette werden beispielsweise sowohl von Genen kodiert, die direkt in den Mitochondrien als auch im Kern liegen. Aber es gibt auch Ausnahmen: Kompex II der Atmungskette wird beispielsweise ausschließlich im Kern kodiert [2]. Kein Wunder also, dass es hier ebenfalls Problemen kommen kann.
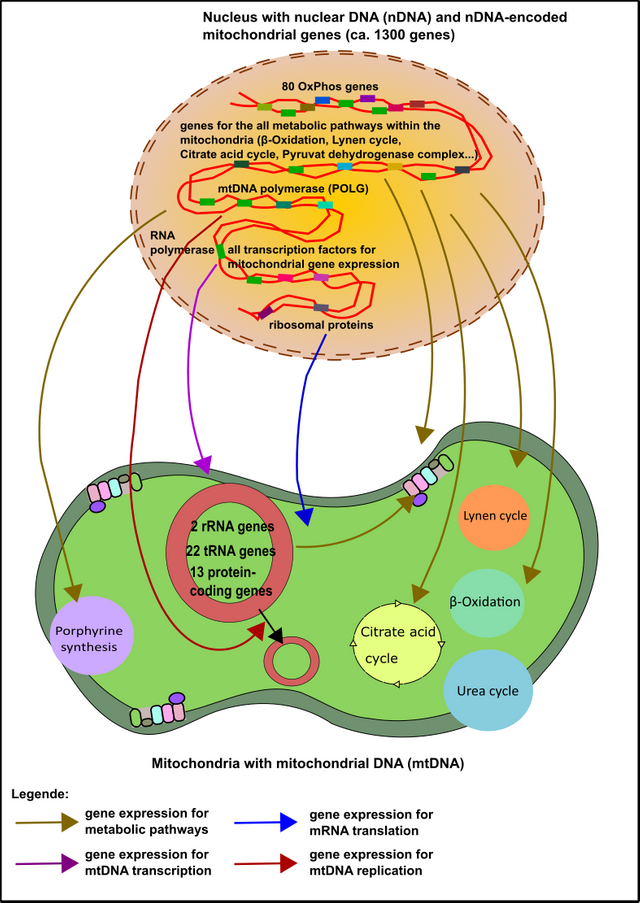
Abbildung 7: Die Mehrheit der mitochondrialen Gene sind im Kern kodiert. Diese „Kerngene“ sind für alles notwendig, sowohl für sämtliche Enzymatik, als auch für die DNA-Replikation des mitochondrialen Genoms (mtDNA). Darüber hinaus arrangieren die im Kern gespeicherten Gene die Transkription der mtDNA-Gene und die Translation der mRNAs, welche die mtDNA hervorbringt. Made by Chapper - unrestricted use allowed
Einen Vorteil hat der Kern allerdings: Er ist weniger stark anfällig für Mutationen als die DNA der Mitochondrien! Warum? Nun so richtig weiß das niemand. Sicherlich spielt es eine entscheidende Rolle, dass Mitochondrien die Nummer 1-Adresse für die Entstehung von Sauerstoffradikalen & Co. bzw. oxidativen Stress (Reaktive Oxygen Spezies, ROS) sind [17-19]. Da die Mito-DNA (mtDNA) direkt daneben liegt, ist es klar, dass diese umso schneller mutiert wird. Außerdem ist das Mitochondriengenom viel kleiner, d.h. wenn eine ROS vorbeikommt, dann trifft sie umso schneller auch ins Schwarze. Bedenkt bitte, dass das Mitochondrien-Genom fast durchgängig mit kodierenden Sequenzen voll ist, die Kern-DNA hingegen besteht zu über 98% aus jenem Material was nicht-kodierend ist [20]. Vieles davon ist landläufig als Junk, also Müll bekannt ist (zu Unrecht übrigens) [20, 21].
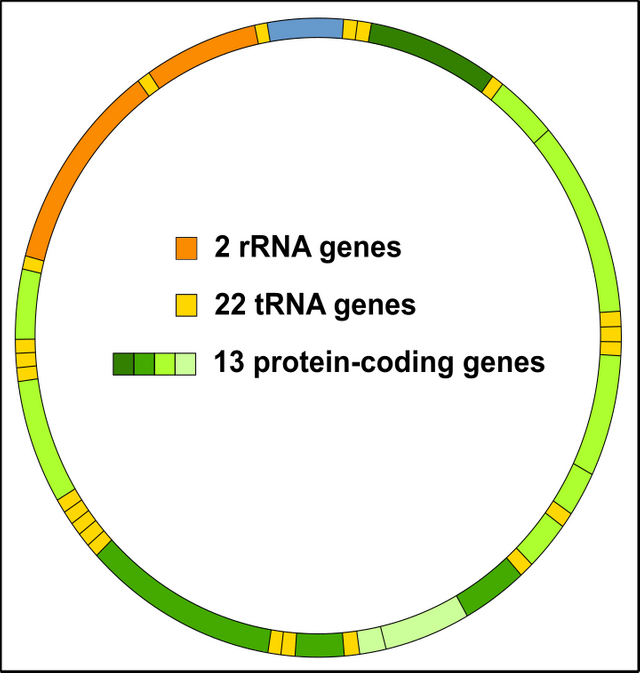
Abbildung 8: Das Mitochondriengenom (mtDNA) ist dichtgepackt mit Genen. Jeder Schuss ist somit ein Treffer! Lizenzfrei CC BY-SA 3.0 modifiziert von Chapper.
{kind=link}
Ihr habt also im Kern viel weniger ROS auf vielmehr DNA (Mito-DNA ca. 1600 bis 1700 Basenpaare vs. Kern-DNA ca. 2.000.000.000 Basenpaare) und darüber hinaus gleicht die Zerstörung von Mito-relevanten Genen durch ROS in der Kern-DNA der Suche nach der berühmten „Nadel im Heuhaufen“ [1, 2, 9, 12, 20, 21]. Wallace 2010 [2] ist übrigens der Meinung, dass der Zelle die mtDNA völlig egal ist, weil in der Natur fehlerhafte Mitos eh keine Chance haben und es eher auf Vermehrung statt Reparatur ankommt. Letztere ist viel zu teuer und wenn ein Organismus sich nicht permanent bei McDonalds und Burger King den nächsten mTOR-Boost abholen kann, werden fehlerhafte Mitochondrien sowieso meist sofort eliminiert. Allerdings hat die Zelle da wohl die Rechnung ohne die moderne Gesellschaft gemacht.
Aber ich schweife ab, denn es gibt durchaus Mutationen in mitochondrialen Genen, welche im Kern liegen. Das Spektrum reicht von schwerwiegenden Krankheiten wie dem Leigh-Syndrom, welches bereits im Kindesalter tödlich verläuft, bis hin zu Depressionen [1, 2]. Das Spektrum ist vielfältig. Auch hier ist prinzipiell keine wirksame therapeutische Maßnahme bekannt.
Kommen wir also zu Punkt 5. und brechen allmählich in die große weite Welt der Pathways auf: Crosstalk zwischen Mitochondrien und Zellkern.
Wie ihr wisst stehen Kern und Mitochondrien in einem permanenten Austausch miteinander. Dies resultiert nicht nur aus dem Fakt, dass die Mitochondrien den Großteil ihrer genetischen Information einst dorthin exportierten [16]. Es ist vielmehr eine zwangsläufige Tatsache, denn die Mitochondrien beeinflussen das Zellgeschehen über Syntheseleistungen, Bereitstellung von Energie, Regulation der Ionenkonzentration bis hin zur Entscheidung über Leben und Tod [11, 12]. Ein Crosstalk zwischen Kern und Mitochondrien ist daher absolut entscheidend, denn andererseits funktioniert das gesamte zelluläre Gefüge nicht.
Aber wie können diese nun miteinander ins Gespräch kommen?
Zunächst natürlich dadurch, dass der Kern die Bildung von frischen Mitochondrien veranlasst.
Dies wird unter anderem durch den Co-Aktivator PGC1α realisiert [22]. Der geneigte Leser erinnert sich bestimmt, dass Transkriptionsfaktoren immer jene Proteine waren, welche die RNA-Polymerase bei der Suche von bestimmten Genen, sowie deren effizienter Ablesung unterstützten (schaue er hier nochmal nach!). Co-Aktivatoren sind quasi jene Faktoren, welche den Transkriptionsfaktoren behilflich sind, damit diese wiederum der RNA-Polymerase besser helfen können [23]. PGC1α wird jedenfalls unter Bedingungen wie Kälte oder Hunger, aber auch nach dem Sport oder im Zuge von oxidativem Stress aktiviert [22]. Danach wandert PGC1α in den Zellkern und ermöglicht hier die Synthese von Genen, welche diesen „Problemen“ entgegenwirken. Hunger kann zum Beispiel durch die vermehrte Verwertung von Fettsäuren kompensiert werden. Dies ist aber wie bereits erwähnt nur in den Mitochondrien möglich (also zumindest die volle Energiegewinnung, Fettsäuren können auch in den Peroxisomen abgebaut werden) [11, 12]. Friert die Zelle kann diese durch das sogenannte Uncoupling Protein (UCP), den Protonengradienten der Mitochondrien entkoppeln und statt ATP einfach Wärme bilden [24]. Oxidativer Stress kommt vor allem aus kaputten Mitochondrien, höchste Zeit neue zu bauen. PGC1α gibt außerdem den entscheidenden Impuls zur Bildung vieler Schlüsselenzyme, welche dem oxidativen Stress entgegenwirken (erinnert euch nur an das Glutathion-System, hier) [22]. Somit setzt der Kern die erste Grundlage für eine fruchtbare Kooperation (Abb.9).
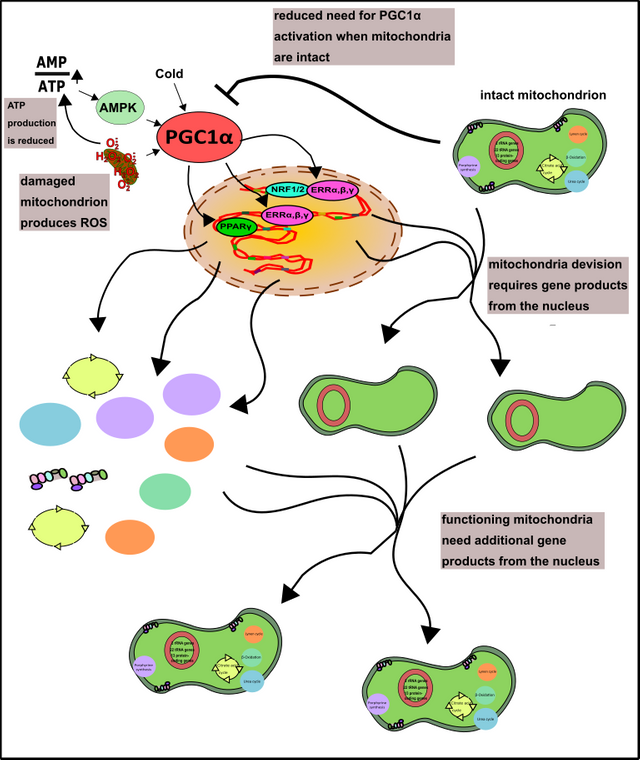
Abbildung 9: Die Rolle von PGC1α in der Bildung von Mitochondrien. Die häufig Stress-assoziierte Aktivität von PGC1α kann durch gut funktionierende Mitochondrien vermindert werden. AMPK habe ich übrigens hier erklärt! Made by Chapper - unrestricted use allowed
Weiterhin stellt der Kern zusätzliche Produkte und Pathways bereit, welche um die Mitochondrien herum wirken, wodurch deren Betrieb erst möglich wird. Dies sind beispielsweise das Zytoskelett in welchem die Mitochondrien eingebettet sind [25] oder auch die Produkte der Glykolyse, Amino- und Fettsäuren. All diese Produkte dienen nicht nur der Energiegewinnung, die letztlich auch dem Kern wieder zu Gute kommt, diese Produkte sind natürlich auch Vorstufen von Bestandteilen der Mitochondrien selbst (Proteine, Membranen etc.) [11, 12, 26, 27].
Die Mitochondrien ihrerseits regulieren die Funktionen im Kern durch ihre Stoffwechselprodukte.
Die Wirkung dieser Produkte ist vor allem epigenetischer Natur [28]. Das heißt, dass sie nicht direkt den Code verändern, sondern die DNA oder deren assoziierte Proteine modifizieren. Durch diese Mechanismen können die Mitochondrien wiederum effektiv die Versorgung mit bestimmten Genprodukten erhöhen, aber auch vermindern. Verantwortlich dafür sind vor allem Zwischenprodukte des Zitratzyklus wie α-Ketoglutarat, Fumarat und Succinat, aber auch Acetyl-CoA,. Weiterhin können auch die schon häufiger erwähnten ROS epigenetische Modifikationen vornehmen und dadurch Änderung der Beziehung zwischen Kern und Mitochondrien bewirken (Abb.10).
Der Kern kann seinerseits aber auch die Mitochondrienleistung drosseln.
Beispielsweise, wenn aufgrund von erhöhtem oxidativem Stress die Autophagie ausgelöst wird, um die ROS-produzierenden Mitochondrien abzuverdauen [4]. Außerdem kann der Kern auch die Synthese von Mitochondrien oder mitochondrialen Produkten unterbinden, um einer überschießenden Energieproduktion entgegenzuwirken [29].
Weiterhin kommt es bei Unterversorgung mit Sauerstoff zu Induktion von HIF1 (Hypoxia-inducible factor 1) [12]. Dies passiert unter anderem in euren Muskeln, wenn ihr Sport treibt [11, 12, 18, 30, 31]. Durch eine Unterversorgung von Gewebe mit Sauerstoff (Hypoxie) kommt es zur vermehrten ROS-Produktion (u.a. aufgrund des vermehrten Abbaus von AMP) [18]. Glücklicherweise gibt es HIF1, welches die Mitochondrien ab- und die Glykolyse dafür anschaltet (bleiben die Mitochondrien an, fressen die auch den letzten Sauerstoff noch weg) [32]. Der Mechanismus schließt die erhöhte Bildung von Glucosetransportern (GLUT1) und die vermehrte Expression der Lactat-Dehydrogenase mit ein (ihr erinnert euch in dem Zusammenhang bestimmt noch an meinen Kombucha-Artikel). Die Mitochondrien werden unter anderem durch die vermehrte Aktivität der PDK1 abgeschaltet. PDK1 ist eine Proteinkinase, welche die Pyruvat-Dehydrogenase in den Mitochondrien inhibiert (mehr zu Proteinkinasen könnt‘ ihr hier lesen). Dadurch kann zumindest schonmal keine Glucose mehr über die Mitochondrien weiter verstoffwechselt werden (keine aerobe Glykolyse mehr möglich, somit wird auch kein wichtiger Sauerstoff mehr verbraucht). Dies ist unter anderem der Grund warum Leistungssportler oft auf „Schnelle Kohlenhydrate“ schwören, denn Glykolyse (hier die anaerobe Glykolyse) ist wie wir bereits wissen sehr ineffizient (2 Mol ATP pro Mol Glucose vs. >30 Mol ATP) und daher brauchen die Sportler reichlich Zucker um ihren Bedarf an ATP ausreichend decken zu können. Alternativ ist natürlich auch Phospho-Creatin eine Option [31].
Wichtig ist einfach, dass man die Akkumulation von AMP vermindert, nicht nur weil dadurch ROS entsteht, sondern, weil es mehrere Tage dauern kann, bis wieder ausreichend ADP für die ATP-Produktion zur Verfügung steht (Abb.10) [31].
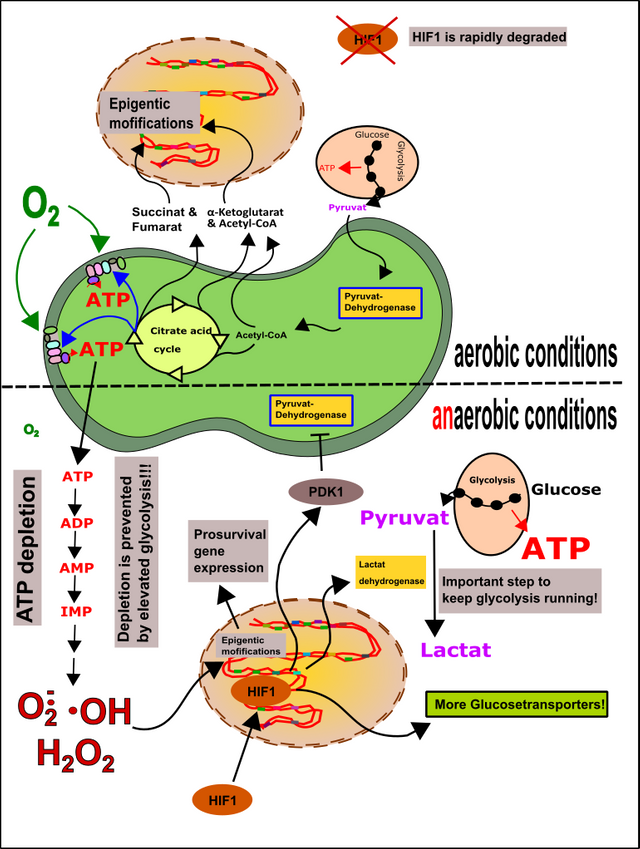
Abbildung 10: Epigenetik und HIF1. Metabolite der Mitochondrien, sowie ROS können epigenetische Modifikationen in der Kern-DNA bewirken und so das Schicksal der Zelle formen. HIF1 wird bei ausreichender Sauerstoffversorgung schnell abgebaut. Bei Sauerstoffmangel wandert HIF1 in den Zellkern und bewirkt in der Endkonsequenz das Abschalten der Mitochondrien und die Verstärkung der Glykolyse. Ziel ist es weiteren Sauerstoffverbrauch zu minimieren und ausreichend ATP zu generieren, damit der Abbau von ATP gestoppt und die Bildung von ROS verhindert wird. Made by Chapper - unrestricted use allowed
Im Ernstfall muss die Zelle sterben.
Letztlich muss sich die Zelle auch darauf verlassen können, dass beispielsweise bei schwerwiegenden DNA-Schäden (welche die Zelle entarten lassen könnten) der programmierte Zelltod (Apoptose) ausgelöst wird [12]. Dies wird durch den Transkriptionsfaktor p53 bewirkt, welcher u.a. die Bildung des Proteins Bax bewirkt. Bax seinerseits bildet in der Membran der Mitochondrien einen Kanal, welcher die Freisetzung von Cytochrom c (eines der Elektronentransportproteine in Atmungskette) auslöst. Cytochrom c ist wiederum wichtig für die Errichtung des sogenannten Apoptosoms, welches anschließend den gesamten Zellinhalt zerschreddert.
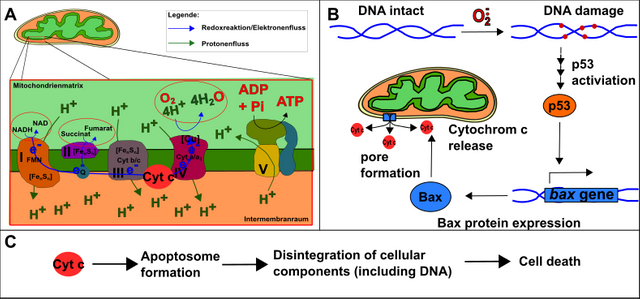
Abbildung 11: Grundlage des programmierten Zelltods (Apoptose). A. Die Atmungskette mit Cytochrom c. B. DNA-Schäden hervorgerufen z.B. durch ROS bewirken die Aktivierung von p53. P53 bewirkt die Bildung von Bax, welches einen Kanal in Mitochondrienmembran bildet. Dadurch kann Cytochrom c aus der Atmungskette „entkommen“. C. Cytochrom c bildet mit weiteren Faktoren das Apoptosome, welches den Zelltod vollzieht. Made by Chapper - unrestricted use allowed
Ich denke die kleine Auswahl reicht (und in Wahrheit sind es hunderte, wenn nicht gar tausende Faktoren, die mitwirken), um euch klar zu machen, dass es noch zahlreiche weitere Möglichkeiten gibt, welche die optimale Funktion der Mitochondrien in eurem Körper torpedieren können. Alles dies wirkt sich natürlich im Großen auf eure Leistungsfähigkeit aus und kann im schlimmsten Falle zu solchen Erkrankungen wie beispielsweise Krebs führen [2, 4, 12, 33]. All diese Wechselwirkungen setzen natürlich funktionierende Mitochondrien voraus und daher ist klar, dass bei nicht-optimaler Erfüllung dieser Voraussetzungen durchaus die Bezeichnung mitochondriale Dysfunktion angebracht ist.
Vom letzten Beispiel ist es natürlich nur ein Katzensprung zu Punkt 6.: Crosstalk mit anderen Zellorganellen.
Dies ist im Wesentlichen noch ein sehr junges, aber womöglich auch gehaltvolles Feld. Ich will mich daher nur basal damit befassen. Jüngst wurde von Hirabayashi et al. [34] herausgefunden, dass ein bestimmtes Protein namens PDZD8 in der Membran des Endoplasmatischen Retikulums (ER) dafür verantwortlich ist, die Mitochondrien mit dem ER in Kontakt zu bringen. Das ER ist nicht nur wichtig für die Synthese von Cholesterin und Proteinen, es dient auch als Calciumreservoir [11, 35]. Calcium seinerseits ist ein entscheidendes Signalmolekül und Cofaktor von verschiedenen Enzymen. In Nervenzellen werden Signale von Zelle zu Zelle unter anderem durch Aufnahme von Calcium ermöglicht. Die erhöhte Calciumkonzentration im Zellplasma wird anschließend durch das ER und die mit ihm verbundenen Mitochondrien konstant erhalten! Haut dieser Verbindung nicht ganz hin, ist die Signalleitung in den Nervenzellen möglicherweise gestört [35].
Doch auch andere Organellen wie die Lysososmen interagieren mit den Mitochondrien. Zum einen natürlich aufgrund ihres Beitrags zum Aufbau unseres zellulären Packmans (siehe dazu hier). Zum anderen aber auch durch die Freisetzung von bestimmten Proteinen, wie von Cathepsinen. Cathepsine ihrerseits können das Protein Bcl-2 inhibieren, welches Bax inhibiert [36]. Dadurch spielen die Lysosomen eine mögliche Rolle im programmierten Zelltod.
Und es geht noch weiter, denn viele andere Einflüsse wirken auf die Mitochondrien ein (Punkt 7.)
Ich will mich hier aber kurz fassen und lediglich darauf hinweisen, dass durch die Abgabe zahlreicher Substanzen aus den Mitochondrien, der Beeinflussung zahlreicher Gene, sowie der Modifikation zahlreicher Signalmoleküle auf diesem Wege, auch zahlreiche Änderungen in den Mitochondrien wieder ankommen. Klingt kompliziert? Ja das ist es auch, aber dieses Geflecht ist nun mal die Grundlage aller Reaktionen eures Körpers. Ich entferne mich hier bewusst von all den Stoffwechselwegen, die wir schon zur Genüge behandelt haben. Auch gehe ich nicht schon wieder auf die Autophagie ein, denn unser zellulärer Packman sollte allmählich sprichwörtlich in euer aller Fleisch und Blut übergegangen sein. Auch will ich nicht auf irgendwelchen Genexpressionen, Epigentics oder ROS rumreiten. All diese Dinge sind für sich genommen schon enorm komplex und lediglich die Spitze des Eisbergs. Das von mir oft als Zelluläres Gezwitscher bezeichnete Rauschen aus Abermillionen von Molekülen, die in feinstem Konzert euer aller Gleichgewicht bestimmen, ist penibel justiert und daher auch stark anfällig. Klar, dass Ungleichgewichte hier, wiederum eure Mitochondrien und somit das gesamte Gefüge durcheinanderbringen können.
Punkt 7 wäre somit ein enorm umfangreiches neues Kapitel, der Artikel ist aber schon wieder viel zu lang geworden, deshalb belasse ich es an dieser Stelle einfach mal dabei.
Ich denke jedem ist klar, dass man mitochondriale Dysfunktionen eher als „zelluläre Dysfunktionen“ bezeichnen sollte.
Den Kern dieser zellulären Dysfunktionen, stellen aber die selbst Mitochondrien dar! Eine einseitige Fokussierung nur auf die Mitochondrien ist allerdings zu kurzgefasst, weil viele (entscheidende) Randbereiche unbeachtet bleiben.
Ihr seid keine Mitochondrien auf zwei Beinen!
Wir fassen also zusammen:
Mitochondrien sind die Drehscheibe des zellulären Geschehens (ich sage bewusst nicht Stoffwechsels). Die DNA der Mitochondrien (mtDNA) ist dichtgepackt mit Genen, welche durch in den Mitochondrien produzierte ROS, schnell mutieren können. Manche dieser Mutationen haben sich als Haplotypen in geographisch klar definierbaren Populationen erhalten und stellen für jene einen Selektionsvorteil dar. Andere Mutationen kommen erst „kürzlich“ vor und können schwerste Krankheiten wie MELAS, MERRF oder LHON hervorrufen. Da viele Krankheiten sich im Laufe des Lebens verschlimmern ist fraglich, ob nicht eine gezielte Selektion auf womöglich noch vorhandene intakte Mitochondrien sinnvoll ist. Gegenwärtig hofft man allerdings eher auf den „Durchbruch der Gentherapie“. Dabei ist der Ansatz bezüglich „Mitochondrienselektion“ nicht abwegig, denn die Mitophagie kaputter Mitochondrien (Autophagie der Mitochondrien) ist eine wissenschaftlich gesicherte Tatsache.
Aber auch weitere Faktoren in und um die Mitochondrien müssen untersucht werden, um eine sinnvolle Therapie anzubieten, dadurch werden die mitochondrialen Dysfunktionen wesentlich weitergefasst und können eher als zelluläre Dysfunktionen bezeichnet werden. Als Beispiel sei hier noch die Krankheit Porphyria variegata genannt. Bei Porphyria variegata (oder einfach Porphyrie) handelt es sich um Störungen in der Porphyriensynthese, welche zum Teil in Mitochondrien abläuft (die letzten beiden Schritte) [33]. Da Porphyrine unter anderem wichtig für den Sauerstofftransport in den roten Blutkörperchen sind, kann so eine Erkrankung schon recht problematisch werden. Die Symptome reichen über Juckreiz, Schmerzen, Muskelschwäche bis hin zu schweren neurologischen Ausfällen [37]. Man glaubt heute, dass sowohl König Georg III. von England als auch Graf Dracula (Vlad III.) an sowas litten. Dies würde jedenfalls die katastrophale Diplomatie seitens Englands erklären, welche letztlich in den Unabhängigkeitskrieg und somit zur Gründung der USA führte. Auch die Leidenschaft des Pfählens seitens Graf Dracula könnte durch „vorrübergehende“ psychische Probleme, verursacht durch eine Porphyrie, erklärt werden. So sehen es jedenfalls manche Wissenschaftler heute [37].
Abb. 12: Dracula (CC0, from Pixabay), Mechanismus der Sauerstoffbindung und Flagge der USA (beides von Chapper).
Man kann die ganze Sache also endlos weiterspinnen und hat dann (wie ich jetzt gerade) keine Zeit mehr sich um Diagnosemöglichkeiten, Therapie und Beratung zu kümmern. Das holen wir aber nach, denn ich werde noch einen dritten Teil (sollten ja erst nur zwei werden) zusammenbasteln.
Bis dahin
Passt auf euch auf und
May Da Mitos Be With You.

Euer Chapper
Achja, fast hätte ich es vergessen:
Der Titel der Serie stellt ja die Frage ob Mito-Medizin ein Trend oder eine Tatsache sei. Ich will es mal so ausdrücken: Während ihr diese Zeilen lest befinden sich ca. 100 g ATP in eurem Körper. Innerhalb eines Tages bildet ihr aber bis zu 100 kg davon [31]. Kombiniert mit den schier endlosen Fakten, welche wir heute hier zusammengetragen haben, sollte klar sein, dass die Mitochondriengesundheit (und alles was dazu gehört) eine wissenschaftliche Tatsache ist.
Quellen:
- Suomalainen, A. and B.J. Battersby, Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat Rev Mol Cell Biol, 2018. 19(2): p. 77-92.
- Wallace, D.C., Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen, 2010. 51(5): p. 440-50.
- Kirches, E., LHON: Mitochondrial Mutations and More. Curr Genomics, 2011. 12(1): p. 44-54.
- Hill, B.G., et al., Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem, 2012. 393(12): p. 1485-1512.
- Brand, M.D. and D.G. Nicholls, Assessing mitochondrial dysfunction in cells. Biochem J, 2011. 435(2): p. 297-312.
- Myhill, S., N.E. Booth, and J. McLaren-Howard, Chronic fatigue syndrome and mitochondrial dysfunction. Int J Clin Exp Med, 2009. 2(1): p. 1-16.
- Michalak, K., et al., Treatment of the Fluoroquinolone-Associated Disability: The Pathobiochemical Implications. Oxid Med Cell Longev, 2017. 2017: p. 8023935.
- Lewis, R., Gentherapie, zweiter Anlauf. Spektrum der Wissenschaft Spezial Biologie Medizin Hirnforschung, 2016. 2/2016.
- Ehler, E., et al., AmtDB: a database of ancient human mitochondrial genomes. Nucleic Acids Res, 2019. 47(D1): p. D29-D32.
- McWilliams, T.G. and A. Suomalainen, Mitochondrial DNA can be inherited from fathers, not just mothers. Nature, 2019. 565(7739): p. 296-297.
- Müller-Esterl, W., Biochemie: Eine Einführung für Mediziner und Naturwissenschaftler. 2004: Spektrum Akademischer Verlag.
- Püschel, Taschenlehrbuch Biochemie. 2011: Thieme Verlagsgruppe.
- Metaxakis, A., C. Ploumi, and N. Tavernarakis, Autophagy in Age-Associated Neurodegeneration. Cells, 2018. 7(5).
- Laplante, M. and D.M. Sabatini, mTOR signaling at a glance. J Cell Sci, 2009. 122(Pt 20): p. 3589-94.
- Kast, B., Der Ernährungskompass: Das Fazit aller wissenschaftlichen Studien zum Thema Ernährung - Mit den 12 wichtigsten Regeln der gesunden Ernährung. 2018: C. Bertelsmann Verlag.
- Embley, T.M. and W. Martin, Eukaryotic evolution, changes and challenges. Nature, 2006. 440(7084): p. 623-30.
- Mari, M., et al., Redox control of liver function in health and disease. Antioxid Redox Signal, 2010. 12(11): p. 1295-331.
- Kalyanaraman, B., Teaching the basics of redox biology to medical and graduate students: Oxidants, antioxidants and disease mechanisms. Redox Biol, 2013. 1(1): p. 244-57.
- Naviaux, R.K., Metabolic features of the cell danger response. Mitochondrion, 2014. 16: p. 7-17.
- Palazzo, A.F. and T.R. Gregory, The case for junk DNA. PLoS Genet, 2014. 10(5): p. e1004351.
- W. Janning, E.K., Genetik: Allgemeine Genetik - Molekulare Genetik - Entwicklungsgenetik. 2004: Georg Thieme Verlag.
- Austin, S. and J. St-Pierre, PGC1alpha and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci, 2012. 125(Pt 21): p. 4963-71.
- Lin, J., C. Handschin, and B.M. Spiegelman, Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab, 2005. 1(6): p. 361-70.
- Chen, W., Q. Yang, and R.G. Roeder, Dynamic interactions and cooperative functions of PGC-1alpha and MED1 in TRalpha-mediated activation of the brown-fat-specific UCP-1 gene. Mol Cell, 2009. 35(6): p. 755-68.
- Kuznetsov, A.V., et al., Cytoskeleton and regulation of mitochondrial function: the role of beta-tubulin II. Front Physiol, 2013. 4: p. 82.
- Mileykovskaya, E. and W. Dowhan, Cardiolipin membrane domains in prokaryotes and eukaryotes. Biochim Biophys Acta, 2009. 1788(10): p. 2084-91.
- Horvath, S.E. and G. Daum, Lipids of mitochondria. Prog Lipid Res, 2013. 52(4): p. 590-614.
- Matilainen, O., P.M. Quiros, and J. Auwerx, Mitochondria and Epigenetics - Crosstalk in Homeostasis and Stress. Trends Cell Biol, 2017. 27(6): p. 453-463.
- Cagin, U. and J.A. Enriquez, The complex crosstalk between mitochondria and the nucleus: What goes in between? Int J Biochem Cell Biol, 2015. 63: p. 10-5.
- al., G.e., Taschenlehrbuch Physiologie. 2. Auflage ed. 2015: Thieme.
- Booth, N.E., S. Myhill, and J. McLaren-Howard, Mitochondrial dysfunction and the pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Int J Clin Exp Med, 2012. 5(3): p. 208-20.
- Okamoto, A., et al., HIF-1-mediated suppression of mitochondria electron transport chain function confers resistance to lidocaine-induced cell death. Sci Rep, 2017. 7(1): p. 3816.
- Peter Karlson, D.D., Jan Koolman, Georg Fuchs, Wolfgang Gerok, Ruth Hammelehle, Karlsons Biochemie und Pathobiochemie. Vol. Auflage: 15. 2005: Thieme.
- Hirabayashi, Y., et al., ER-mitochondria tethering by PDZD8 regulates Ca(2+) dynamics in mammalian neurons. Science, 2017. 358(6363): p. 623-630.
- Lombardi, A.A. and J.W. Elrod, Mediating ER-mitochondrial cross-talk. Science, 2017. 358(6363): p. 591-592.
- Repnik, U. and B. Turk, Lysosomal-mitochondrial cross-talk during cell death. Mitochondrion, 2010. 10(6): p. 662-9.
- Hill, F.E.D.E.D., Porphyrine: Ein Ring für die Farben des Lebens. Spektrum der Wissenschaft Biologie Medizin Hirnforschung, 2019. 1/19.

Sehr interessantes Thema und dein Artikel ist wie immer sehr informativ und umfangreich.
Muss ich mir nochmal in Ruhe durchlesen.
Eine kleine Frage hab ich schon, und zwar ist doch der oxidative Stress von Sport nicht schädlich für unsere Mitochondrien oder?
Du hattest ja sogar mal den Tipp gegeben unmittelbar nach dem Sport keine starken antioxidantien zu sich zu nehmen, wenn ich mich nicht irre.
Grüße
Dein Output ist echt beneidenswert. Was soll ich da noch groß sagen? Resteem.
Hi @sco, danke fürs Resteemen und die lieben Worte. Um ehrlich zu sein hilft mir dies meine Gedanken zu ordnen, wenn ich mich mit einem Thema befasse. Du kennst das bestimmt man liest und liest und liest und liest und letztlich bleibt nur ein komisches Drücken in Hirn zurück. Die Sachen einfach mal runterzubrechen hilft mir diese Blockaden zu lösen. Jedes Mal habe ich das Gefühl ein Stück weitergekommen zu sein und außerdem habe ich das Wichtigste zusammengefasst irgendwo liegen und kann bei Bedarf schnell drauf zugreifen. Win/Win quasi!
Posted using Partiko Android
Kenn ich sehr gut. Ich nehm mir seit Ewigkeiten vor wieder zu bloggen, aber irgendwie wird's momentan einfach nichts. Und die Gedanken schwirren.
Ich habe mir da sone Excel-Tabelle angelegt wo alles reinkommt was ich so interessant finde. Für's erst reicht das um den "Brainfog" etwas Herrr zu werden.
Grüßle
First of all, you are quite skilled at bridging the gap between the science and lay communities. Quite a trick, writing so you hold the attention of one and bring understanding to the other. Thanks for sharing your extraordinary expertise with us.
I'd like to tell you how very interesting this was to me on a number of fronts. I've been reading up on Huntington's, and as I worked my way through your article began to recognize some information that coincided with research I've come across. Even though much of the technical discussion can get away from me, enough of it gets through to make the whole picture sensible.
The most enjoyable part of your blog is the way you strive to make positive use of your research. This is not a dry, academic endeavor for you. This is the path of hope, of possible cures and a way to better lives. That is most admirable.
I did very much enjoy reading this, and will revisit it. No doubt, it will help me to understand my reading on Huntington's, which I return to on and off (between art projects--after all, without fun, what is life?)
With great respect, I wish you peace and a productive week,
AG
Hey @agmoore, thanks for the kind reply again.
I have to admit that this time the article was pretty complex and much more hard then usually. The reason was just that I recognized the topic is so multi-faced it was a prerequisite to writing them in that way for preventing conceptional errors.
The last part I have to try to bridge the gap for more understandable content. It nice that I have included the most important facts in the first two article thus I just have to refer to them.
May I ask you where your interest in Huntington disease is coming from. Actually, this topic is quite interesting. I read an article about this where the researchers compared various animals. They found that amount these glutamine repeats increased with the evolutionary level of the animals. Very primitive animals (without a brain) had no repeats. Flies and stuff just had one or two. Sea stars had some more and finally mammals, especially humans had many more. Today it seems like that the more glutamine repeats someone has, the better are the neurological abilities (intelligence). Unfortunately, with the bad side effects.
Nevertheless, by knocking out the gene you can interrupt brain formation.
Huntington is, therefore, a "mutation" which is beneficial and destructive at the same time.
Have you ever wrote something about it?
Anyway
I go the bed now.
See ya next time
Chapper
I've been nibbling at the edges of this for a few weeks now. Since I'm not trained in science, I circle around and pick up crumbs until I begin to get a cohesive picture. I've read a few good articles about the relationship between Huntington's, Parkinson's and Alzheimer's. The thrust of the articles seems to be not a cure, but a treatment to modify symptoms. And this seems, in some research, to indicate turning off the signaling that causes the cells to behave in a way that's pathological. I lack the vocabulary to describe what I so far understand, but will acquire that vocabulary in time --I hope. You see why your article was interesting to me. It's one more, really big crumb on the trail :) Here's an article I picked up today, and will save, because of clues laid out in your blog.
I'm pretty sure I'll write about Huntington's in some fashion...but what will I say? Still a mystery to me 😄
This is also the case when you are already trained ;-)
AD & Parkinson is also related to mitochondria. Maybe I will try to go further into this subject in the third part of my series, but I can't promise.
Concerning the Huntington stuff. This is a total interesting case.
Maybe I will write something briefly about it! I wish I would have more time.
Anyway
Hope to read some fresh scientific stuff by you soon.
And don't forget. If I'm not responding it's because of my feed!
So far
All best
Chapper
Thanks for the encouragement. Getting close... Hope to read your blog about Huntington's soon :)
Peace until then,
AG
This post has been voted on by the SteemSTEM curation team and voting trail. It is elligible for support from @utopian-io.
If you appreciate the work we are doing, then consider supporting our witness stem.witness. Additional witness support to the utopian-io witness would be appreciated as well.
For additional information please join us on the SteemSTEM discord and to get to know the rest of the community!
Please consider setting @steemstem as a beneficiary to your post to get a stronger support.
Please consider using the steemstem.io app to get a stronger support.
Hi @chappertron!
Your post was upvoted by Utopian.io in cooperation with @steemstem - supporting knowledge, innovation and technological advancement on the Steem Blockchain.
Contribute to Open Source with utopian.io
Learn how to contribute on our website and join the new open source economy.
Want to chat? Join the Utopian Community on Discord https://discord.gg/h52nFrV
@chappertron You have received a 100% upvote from @intro.bot because this post did not use any bidbots and you have not used bidbots in the last 30 days!
Upvoting this comment will help keep this service running.
Hi, @chappertron!
You just got a 0.3% upvote from SteemPlus!
To get higher upvotes, earn more SteemPlus Points (SPP). On your Steemit wallet, check your SPP balance and click on "How to earn SPP?" to find out all the ways to earn.
If you're not using SteemPlus yet, please check our last posts in here to see the many ways in which SteemPlus can improve your Steem experience on Steemit and Busy.